Multipl Skleroz’un Geçmişi ve Geleceği
Multipl skleroz’un ilk tanımlandığı 40 yıl içerisinde klinik ve patolojik ayrıntıları yeterince aydınlanmıştır. Geçen 120 yılda ise, multipl sklerozun nedeni, inflamatuar demiyelinizasyon ve akson dejeneresansının mekanizmaları konusundaki bilgiler birikmiştir. Geçen 10 yılda, hastalığın seyrini orta derecede etkileyen tedaviler bu alana girmiştir. Yeni araştırmalar ise multipl sklerozun yol açtığı yıkımı sınırlandırmaya, onarıma ve yıkımı önlemeye odaklanmıştır.
NÖROMİYELİTİS OPTİKA (DEVIC HASTALIĞI)
Nöromiyelitis optika (NMO), yakın zaman ilişkisi içerisinde ortaya çıkan optik nöropati ve miyelopati ile nitelenen, pek sık karşılaşılmayan bir nörolojik hastalıktır. Lezyonların optik sinirler ve m. spinalis dışına dağılımı nedeniyle multipl sklerozdan ayrılması bir soru işareti oluşturur. Güçlüğün bir bölümü, geniş, belirsiz ve tutarsız tanımında yatar. Devic hastalığı, Devic sendromu ve nöromiyelitis optika (NMO) sıklıkla biribirinin yerine kullanılır. Devic sendromu yukarıdaki tanıma uyan tüm hastaları kapsamasına karşın, 2. ve 3. tanımların sadece farklı bir bozukluğa sahip olduğu düşünülen hastaları adlandırmada kullanılması yeğlenir. Devic sendromlu hastaların bir bölümünü içine alan optiko-spinal multipl skleroz, başlıca ya da belirgin olarak optik sinir ve m. spinalisin tutulduğu hastaları adlandırmak için kullanılır.
Tablonun patogenez ve fizyopatolojisine göre, Devic sendromu, akut disemine ensefalomiyelit (ADEM) ile, otoimmun hastalıklarla, multipl skleroz ile ve olasılıkla viral infeksiyonlarla birlikte olabilir. Patolojik tanımlamalar çok değişir, çünkü bu hastalar değişmez olarak heterojen bir gruptur.
Klasik olarak, akut m.spinalis lezyonları, yaygın şişme ve yumuşama gösterir, birkaç seviyede uzanır, ya da tüm m. spinaliste devamlı veya yama tarzında dağılır. Akut olarak ak ve gri maddeyi tutan yoğun makrofaj infiltrasyonu, miyelin ve akson kaybı ile damarların etrafında lenfosit kümelerinin oluşmasıyla giden bir yıkım söz konusudur. Kronik lezyonlarda, m. spinalis nekrotik dejenerasyon ve gliozis nedeni ile nekrotik ve atrofiktir. Bu geniş lezyon infarkta benzeyebilir. M. spinalisteki ani şişme, sınırlayıcı pia ile karşılaşınca intramedüller basıncı arttırabilir ve küçük parankimal damarların kapanmasına yol açarak daha ileri doku yıkımına yol açabilir. Kalın ve hiyalinize duvarlı damarların proliferasyonu infarkttan sonra ya da diğer yaygın yıkımlarda görülenlere benzer. Daha az fulminant lezyonlar aynı anda oluşabilir ve bunlar inflamatuar demiyelinizasyona daha çok benzer. Optik sinirler, kiasma, bazen serebral hemisferler benzer tarzda hem demiyelinizan lezyonlar, hem nekrotizan lezyonlarla ya da ikisi bir arada olarak etkilenebilir. Multipl skleroz ile NMO arasındaki patogenetik ilişki net değildir. NMO’yu multipl sklerozdan ayırt ettiren serum antikor biyoişaretleyicisi olan ve aquaporin-4’e karşı oluşmuş olan antikorlar olduğu belirlenen NMO-IgG’nin ortaya konması ve yakında klinik kullanıma girmesi yeni ve heyecan yaratıcı bir bulgu olmuştur. Ancak, etyopatogenezdeki yeri ve klinik önemi konusundaki çalışmalar sürmektedir.
Devic sendromu ile ortaya çıkan hastalar farklı bazı seyir tipleri gösterebilir. Hastaların % 35’i monofazik, % 55’i optik sinir ve m. spinalis’e sınırlı ataklarla ve daha nadir olarak tipik multipl skleroz seyrine uymayan ve ataksız çok hızlı ilerleyici bir seyir gösterebilir. Epidemiyoloji ve risk faktörleri gözden geçirildiğinde Devic sendromu, 1 ile 73 arasında değişen yaşlarda ortaya çıkmaktadır. Monofazik Devic sendromunun ortalama başlangıç yaşı 27 iken, yineleyen Devic sendromunun daha ileri yaşlarda (ortalama 43) belirme eğilimi gösterdiği bildirilir. Monofazik Devic sendromunun ağırlıklı olarak her iki cinste de görüldüğü dikkati çekerken, yineleyen tip daha ağırlıklı olarak kadınlarda görülür (3.8/1). Ataklarla giden nöromiyelitis optika sistemik lupus eritematozus başta olmak üzere sıklıkla otoimmun hastalıklarla birliktedir. Bu hastalarda, sıklıkla eritrosit sedimentasyon hızı ve anti-nükleer antikorlar, anti-ds DNA ve antifosfolipid antikorlar gibi otoantikorların yükselmesi gözlenir.
Hastaların 1/3‘ü nörolojik tablonun başlamasından birkaç hafta önce geçirilmiş bir üst solunum yolu infeksiyonu, grip ya da gastroenterit öyküsüne sahiptir. Devic sendromunun öncesinde en sık karşılaşılan infeksiyonlar su çiçeği ve akciğer tüberkülozudur. Bazen de grip ve kabakulak aşısının ardından Devic sendromu gelişebilir. Yakın zamana kadar Devic sendromunun Japonya ve doğu Asya’da daha sık görüldüğü bildirilmiştir; bununla birlikte sıklığı 100.000’de 5 ten azdır. Kliniğimiz materyalinde, demiyelinizan hastalıklar içersinde Devic sendromunun %1 oranında görüldüğü belirlenmiştir.
Devic sendromundaki klinik bulgular ve eşlik eden belirtiler saatler ya da günler içerisinde yerleşen optik nöropati ve miyelittir. Bu bulguların öncesinde başağrısı, bulantı, uyuklama, ateş ve miyalji tabloya eşlik edebilir. Hastaların büyük çoğunluğu iki yanlı optik nöropati geliştirir. Görme kaybına genellikle göz çevresinde ağrı, miyelite ise belli bölgede bel ağrısı ya da radiküler ağrı eşlik edebilir. Lhermitte bulgusu tabloya eşlik edebilir. M. spinalis hasarına bağlı olarak tonik spazmlar ve nöropatik alt ekstremite ağrıları sık karşılaşılan sekellerdir. Ağır nörolojik kayıplar sıktır ve düzelme derecesi değişkendir.
Ayırıcı Tanı
Devic sendromunun ayırıcı tanısı, multipl skleroz, ADEM, akciğer tüberkülozu ve özellikle bağışıklık sistemi baskılanmış kişilerde viral infeksiyonu içerir. Ailede birden fazla hasta birey bulunması halinde bir mitokondriyal hastalık göz önüne alınmalıdır. Ataklarla seyreden nöromiyelitis optika’da eşlik eden bir otoimmun hastalık aranmalıdır.
İncelemelerde, yapısal lezyonları dışlamak ve patolojik süreç konusunda bilgi sağlamak için görüntüleme ön sırada yer alır. Akut dönemde beyin MR’ında optik sinir ve kiasma genişlemesi, T2 ağırlıklı sinyal değişiklikleri ve kontrast madde tutulması görülebilir. Olguların yaklaşık %25’inde serebral ak madde lezyonları görülebilir. M. spinaliste T2 ağırlıklı sinyal artışı sıktır ve genellikle üst servikal bölge lezyonlarını ortaya koyar. Tipik olarak m. spinaliste şişme, birkaç m. spinalis segmentine uzanan sinyal değişiklikleri gösterir (Şekil 8). Bu görünüm m. spinalis tümörüne benzeyebilir. Hastaların 1/3’ünde eritrosit sedimentasyon hızında artma, yarısında pozitif antinükleer antikorlar, nadiren diğer otoantikorlar görülebilir. Sifiliz, Lyme hastalığı ve edinsel bağışıklık sistemi yetmezliği sendromu diğer laboratuvar testleriyle dışlanmalıdır. Akciğer grafisi tüberküloz ve sarkoidozu ayırt etmede yardımcı olabilir. Devic sendromunun değerlendirilmesinde BOS incelemesi esastır ve bazen yinelenmesi gerekebilir. Akut dönemde, multipl sklerozda görülenin tersine Devic sendromlu hastaların önemli bir bölümünde (%83) BOS’ta hücre artışı saptanır. Bazen milimetreküpte 100 hücreyi aşan hücre artışı görülebilir. Nötrofil artışı ve hakimiyetiyle karşılaşılabilir. Bu durum multipl sklerozda görülmez. Protein artışı genellikle sıktır, ve hastaların %41’inde 100mg/dl’yi aşar. Beyin omurilik sıvısındaki bu yoğun inflamatuar yanıta karşın pozitif oligoklonal band olguların %20’sinden azında görülür. Özellikle bağışıklık sistemi baskılanmış kişilerde olmak üzere herpesvirus ailesi için serolojik inceleme yapılması önemlidir.
Şekil 8.Nöromiyelitis optika. Kranyal MR.
Şekil 8A. T2 ağırlıklı sagittal kesitlerde servikomedüller bölgede izlenen hiperintens inflamatuvar demiyelinizan lezyon
Şekil 8B. T2 ağırlıklı sagittal kesitlerde alt dorsal bölgeye uzanan hiperintens inflamatuvar demiyelinizan lezyon
(Resimler, İstanbul Tıp Fakültesi, Nöroloji Anabilim Dalı Multipl Skleroz Birimi materyelinden saglanmıştır).
Tedavi
Akut ve subakut Devic sendromlu hastaların klinik tablosu genellikle intavenöz metilprednizolon gibi kortikosterid tedavilere yanıt verir. İntravenöz metilprednizolon önemli bir iyileşme meydana getirmezse plazma değişimi seçenekler arasında yer alır. Nüksleri ve gelişecek özürlülüğü önleme konusundaki girişimler, bağışıklık sistemini baskılayıcı tedavilerin uygulanması dahil sıklıkla hayal kırıcı olmakla birlikte, yineleyici hastalarda bağışıklık baskılayıcı tedaviler tedavinin esasını oluşturmaktadır.Hastaların izlenmesinde destekleyici tedavi esastır. Bu hastalar, derin ven trombozu, akciğer embolisi, idrar yolu infeksiyonu, dekubitis ülserleri ve kontraktürler gibi birçok komplikasyona açıktır.
Monofazik Devic sendromlu hastalarda optik nöropati ve miyelopati genellikle aynı anda ve hızlı bir şekilde ortaya çıkar (aradaki süre genellikle 1 ile 30 gün arasında değişir). Hastaların bazılarında ciddi sekel kalırken, çoğu çarpıcı şekilde nörolojik kayıpsız düzelir. Özgeçmişteki müphem nörolojik belirti ve bulgular gelecekteki nüksleri, gerek tipik multipl skleroz, gerekse yineleyici nöromiyelitis optika gelişimini tahmin etmek bakımından önemlidir.Yineleyen optik nöropati ve miyelopatisi olan hastalarda optik nöropati ile miyelopati arasında uzun bir süre bulunabilir. Yineleyen nöromiyelitis optikalı hastaların geri kalan büyük çoğunluğu sık ve ağır ataklarla giden kötü bir prognoza sahiptir.
MERKEZİ SİNİR SİSTEMİNİN İZOLE İNFLAMATUAR DEMİYELİNİZAN SENDROMLARI (KİS)
Önceden belirtisi olmayan bir kişide belirli bir bölgeye sınırlı merkezi sinir sistemi demiyelinizasyonu sıklıkla multipl sklerozun ilk belirtisidir. Bununla birlikte bazen silik şekilde bir ADEM olabilir. Optik sinir, m. spinalis ve beyinsapı bu yineleyen tek belirtili olayların en sık karşılaşıldığı bölgelerdir. Bunların patogenezleri, patofizyolojileri, epidemiyolojisi, klinik tablolara eşlik eden bozuklukların ayırıcı tanı, değerlendirme ve hasta yönetimi daha önce tartışılan multipl sklerozdakine benzerdir.
Bu olgularda, optik nöropatinin her bir epizodundan sonra görsel iyileşme için prognoz iyidir, ve hastaların çoğu normal görme keskinliklerini kazanırlar. Otuz beş yaşından sonra yineleyen optik nöropati ve ağır görme kaybı prognozun iyi olmayacağına ilişkin ipuçlarıdır. Araştırıcılar, izole optik nöropati geçirdikten sonra hastalar 20 yıldan daha uzun süre izlendiklerinde, %50 ya da fazlasının KKMS’e dönüştüğü sonucuna varmışlardır. Riskin büyük bölümü ilk birkaç yıla sınırlıdır, buna karşılık önemli risk 4. onyıla kadar sürebilir. Çocuklar daha sıklıkla iki yanlı optik nöropati geçirirler ve erişkinlere göre daha düşük multipl skleroz geliştirme riski taşırlar. Retinal damarlarda venöz kılıf oluşumu, yineleyen optik nöropati, ailede multipl sklerozlu birey bulunması, beyaz ırka mensup olma, önceye ait müphem özgül olmayan nörolojik belirtiler, BOS’ta oligoklonal bandlar ve artmış IgG indeksi ve beyin MRG’sinde ak madde lezyonlarının görülmesi multipl skleroz gelişme riskini arttırır. Bir çalışmada monosemptomatik optik nöropatili 44 hastanın anormal MRG bulguları olan %82’si, 5 yıl içersinde multipl skleroz tanısının tüm ölçütlerini taşır hale gelmiştir. Oysa, MRG’de normal ya da özgül olmayan bulgusu olanların %6’ sı multipl skleroz geliştirmiştir. Optik nöropati tedavi çalışmasında da MRG’de serebral ak madde lezyonları olanlarda multipl skleroz gelişme riski artmış bulunmuştur. Her iki çalışma da lezyon sayısı ne kadar fazla ise multipl skleroz gelişme riskinin o kadar fazla olduğunu bulmuştur.
Akut transvers miyelopatinin ağırlığı daha sonra belirti veren demiyelinizan lezyonların ortaya çıkışıyla ters orantılıdır. Ağır motor zaaf, duyu ve sfinkter kusuru ile giden transvers miyelopatili olgularda ilerde multipl skleroz gelişme riski oldukça düşüktür. Tepe noktasında motor işlevlerin korunmasıyla parsiyel transvers miyelit daha yüksek sıklıkta multipl skleroz ile birliktedir. Monosemptomatik beyinsapı demiyelinizasyonu akut transvers miyelit ve optik nöropati kadar sık değildir. Sınırlı sayıdaki çalışmalardan birinde, MRG’de ak madde lezyonu olan hastaların ¾ ‘ünde izleyen 5 yılda multipl skleroz gelişmiştir.
Belirtildiği gibi klinik izole sendrom (KİS) gösteren hastalar, klinik kesin MS geliştirme bakımından yüksek risk taşırlar. Epidemiyolojik ve klinik çalışmalar KİS hastalarının %85’inin iki yıl içersinde MS geliştirdiğini ortaya koyar. Erken tedavinin, MS’e dönüşme riskini azalttığına ilişkin çalışmalar mevcuttur. Erken aksonal kayıp inflamasyonla birliktedir ve geri dönüşümsüz olabilir. KİS oluşumundan kısa süre sonra atrofi görülebilir. MRG parametreleri de KİS’in MS’e dönüşümü konusunda yol gösterici olabilir. KİS hastaları ortalama 7 yılda yeni T2A lezyonlar geliştirir, ve klinik kesin MS’e dönüşme riskleri artar. Ayrıca, lezyonların hacmindeki artma uzun süreli özürlülüğü artırmaktadır. CHAMPS ve PreCISe çalışmaları monofokal hastalarda, ETOMS ve BENEFIT çalışmaları mono- ve multifokal hastalarda erken tedavinin plaseboya göre MS gelişme riskini istatistiksel olarak anlamlı bir şekilde azalttığını ortaya koymuştur. Diğer önemli bir yön, son yıllarda ilaçların etkinliğinin MS’in fazına göre değiştiğini ortaya koymaktadır. KİS döneminde ilaç etkinliği yaklaşık %50, RRMS döneminde %30 dur ve hastalık seyri ilerledikçe azaldığı bildirilmiştir.
SCHILDER’İN MİYELİNOKLASTİK DİFFÜZ SKLEROZU
Bazen serebrumda tek büyük bir odak ya da birkaç odak şeklinde yaygın demiyelinizasyon ile karşılaşılır. Bu tür lezyonlara erişkinlere göre çocuk ve ergenlerde daha sık rastlanır. Schilder’in orjinal yayınına ve tanımına konu olan 14 yaşındaki kız çocuğunun ilerleyici mental retardasyon ve kafa içi basınç artması sendromu geliştirdiği ve başlangıçtan 19 hafta sonra tablonun ölümle sonlandığı bilinir. Nekropside, her iki hemisferde, ak madde içersinde, büyük ve sınırları belirgin, kapalı demiyelinizasyon alanları görülür. Bunlara multipl sklerozun sık karşılaşılan lezyonlarına benzeyen çok sayıda daha küçük demiyelinizasyon odakları eşlik eder. Lezyonlarda inflamatuvar reaksiyonun belirgin olması ve aksonların nispeten korunmasıyla multipl skleroz lezyonlarına benzemesini dikkate alarak, Schilder bu hastalığa “encephalitis periaxialis diffusa” adını vermiştir. Ne yazık ki, izleyen yayınlarda Schilder farklı tipte iki durumda aynı adı kullanmıştır. Sonraki değerlendirmelerde, bu olgulardan birinde lökodistrofi, diğerinde ise subakut sklerozan panensefalit saptanmıştır. 1979 yılında Ellison ve Baron seyri multipl skleroza benzeyen, tanının biopsi ile doğrulandığı steroide yanıtlı benzer bir olgu bildirmişlerdir.
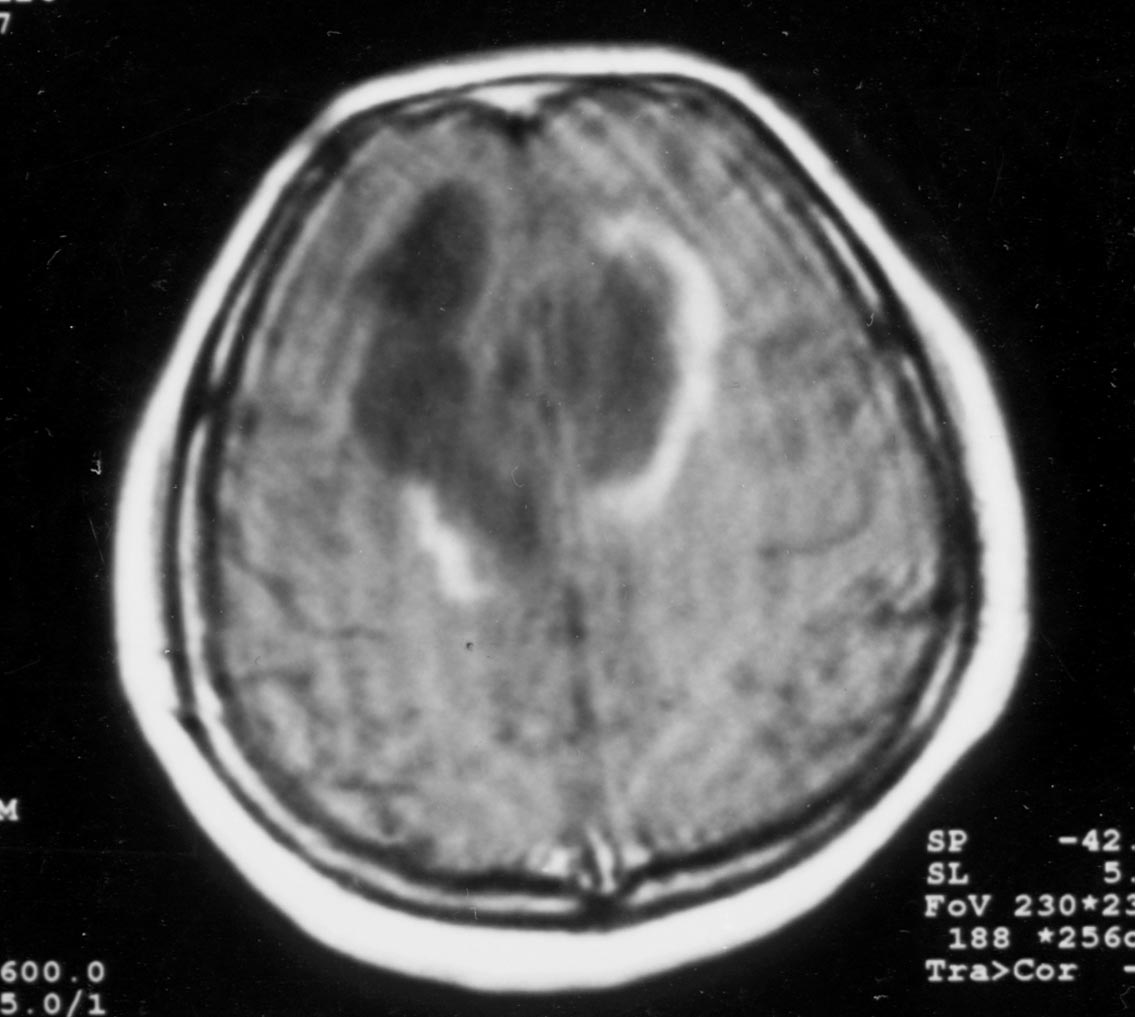
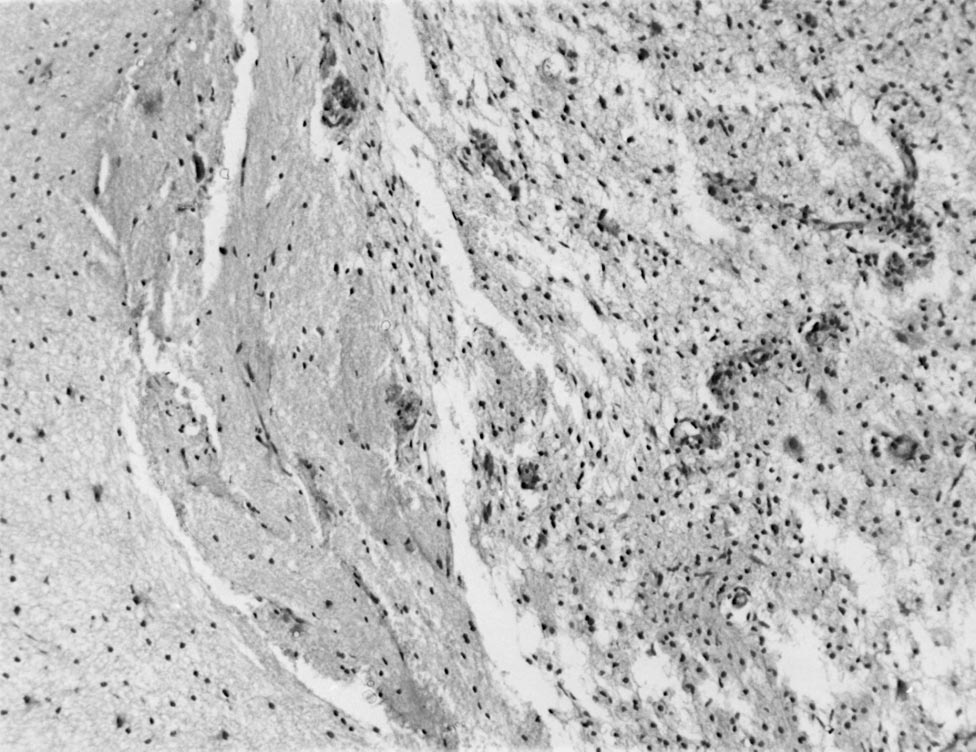
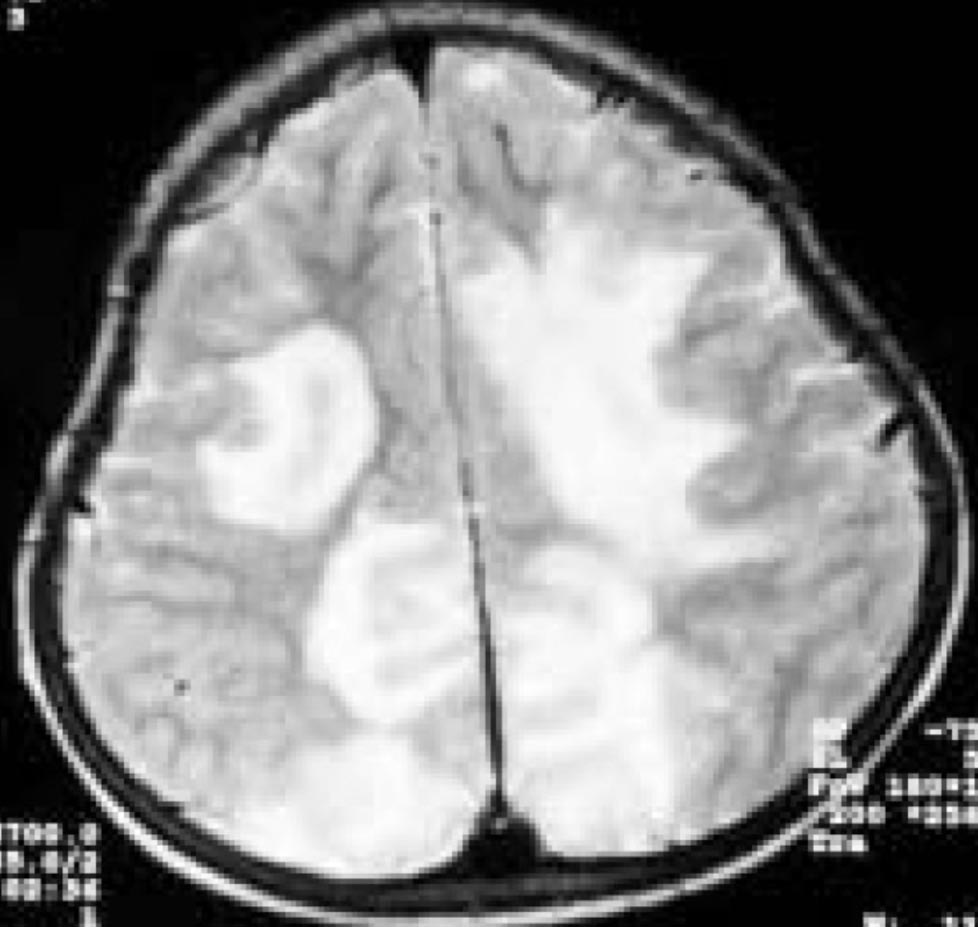
Serebral belirtilerin öncesinde ya da sonrasında optik nöropati gelişebilir, BOS incelemesinde genelikle oligoklonal band negatiftir. Radyolojik olarak öncelikle diffüz serebral neoplazmlardan ayırt edilmeleri gerekir. Bu olgularda tipik lezyon, serebrumda, sayısı 1-3 arasında değişen, büyük, sınırları iyi belirlenmiş, tüm bir lobu ya da serebral hemisferi etkileyen, tipik olarak korpus kallozumu geçerek karşı hemisfere uzanır şekilde yerleşir. MRG’de bu lezyonlar T1 ağırlıklı kesitlerde hipointens, sınırları iyi belirlenmiş ve kistik görünümde olabilir. Çeperinin yoğun ve kalın kontrast tutması onu apse, metastaz vb lezyonlardan ayırt etmemizi sağlar (Şekil 9). Lezyonun hafif-orta kitle etkisi olabilir. T2 ağırlıklı incelemelerde T1’deki lezyon hiperintens olarak izlenir. Neoplazmla karıştırılma sonucu yapılan cerrahi girişimler, steroidle düzelebilecek bu tabloda sekel kalmasına yol açar. Histolojik olarak gerek büyük gerekse küçük lezyonlar multipl sklerozun karakteristik tablosunu gösterir. Poser, literatürü gözden geçirip klinik ve radyolojik tablonun özelliklerini ortaya koyarak, tanı ölçütlerini belirlemeye çalışmıştır. 1986’da konu tekrar gözden geçirilmiştir. Diffüz serebral sklerozun bu tipinin multipl skleroz ile yakın ilişkili göründüğü aşikardır ve Schilder’in öne sürdüğü gibi multipl sklerozun varyantı olabilir.
Şekil 9. Schilder'in miyelinoklastik diffüz sklerozu. Kranyal BT, kranyal MR ve patoloji.
Şekil 9A. Aksiyel BT'de sol parietotemporooksipital bölgeyi tutan, kitle etkisi olan ve kontrast madde verilmesinin ardından iç kenarları halka şeklinde kontrast tutan lezyon (patoloji: inflamatuar demiyelinizasyon).
Şekil 9B. GdDTPA injeksiyonundan sonra aksiyel T1 ağırlıklı kesitlerde, korpus kallosumdan geçerek her iki frontal bölgeye yayılan, hafif kitle etkisi olan, apse ve tümöre göre daha kalın bir halka şeklinde kontrast tutan, tümöre benzer inflamatuar demiyelinizan lezyon (patoloji: C'ye bakınız.)
Şekil 9C. Solda görülen sağlam beyin dokusundan belirgin bir demarkasyon hattıyla ayrılan, daha gevşek görünümlü, yer yer perivasküler inflamasyonun görüldüğü demiyelinizasyon alanı. Patolojik görünüm multipl skleroz plağı ile uyum gösterir. Bu örnek, B'de görülen lezyondan elde edilmiştir.
(Resimler, İstanbul Tıp Fakültesi, Nöroloji Anabilim Dalı Multipl Skleroz Birimi materyelinden saglanmıştır, bu incelemenin sağlanmaslnda emeği geçen Dr. Çiçek Bayındır ve Dr. Orhan Barlas'a teşekkür ederiz).
Tablonun steroide verdiği dramatik yanıt dikkat çekicidir. Bu sayede hastalar daha rahat izlenebilir. Son zamanlarda tablonun yineleyici formları tanımlanmış, ve bu durum Schilder döneminden gelen ağır ve fatal hastalık kavramının sorgulanmasına yol açmıştır.
AKUT DİSEMİNE ENSEFALOMİYELİT (ADEM) ve İLİŞKİLİ HASTALIKLAR
Akut disemine ensefalomiyelit (ADEM), bir döküntülü hastalık veya infeksiyon hastalığının ya da aşılanmanın ardından ortaya çıkan, merkezi sinir sisteminin birçok kısmının eş zamanlı olarak tutulduğu bir ensefalomiyelit tablosudur. Bazı çocukluk çağı infeksiyonlarının ardından post-infeksiyöz ensefalomiyelit gelişebilir. Bunlar arasında boğmaca, kızıl, kızamık, kızamıkçık ve su çiçeği sayılabilir. Özellikle su çiçeği sonrası gelişen ensefalomiyelit tablosu olguların yarısından fazlasında izole bir serebellar sendrom şeklinde karşımıza çıkar ve çok iyi prognoz taşır. Bunların dışında çocuklarda ADEM tablosuna yol açabilen başka ajanlar da tanımlanmıştır: enteroviruslar (ECHO, Coxackie), herpes, mikoplazma ve HIV gibi. Erişkinlerde de non-spesifik üst solunum yolu infeksiyonları, adenoviruslar, mikoplazma ve HIV gibi ajanlar sorumlu tutulmaktadır. Aşılanma sonrasında da ADEM tablosu ortaya çıkabilir. Bunlar arasında bir dönemin önemli sorunlarından biri olan post-vaksiniyal ensefalomiyelit, çiçek aşısının uygulamadan kalkması ile çok nadir görülen bir durum haline gelmiştir. Bir diğer sorun da nöral doku içeren kuduz aşılarından sonra gelişen ensefalomiyelit tablosudur. Bu da tipik ADEM özellikleri gösterir. Yeni tip insan diploid hücre aşısının (HDCV) kullanılmaya başlamasıyla oldukça seyrekleşmiştir. Diğer pek çok aşılanmadan sonra da ADEM gelişebilir. Bunlar arasında hepatit B, kızamık, boğmaca, difteri aşıları sayılabilir. Olguların bir kısmında herhangi bir öncü infeksiyon veya aşı saptanamayabilir.
Belirtiler infeksiyon veya aşılanmadan sonraki birkaç gün ile bikaç hafta içinde ortaya çıkar. Başağrısı ve uyanıklık kusuru sıktır; bazen komaya kadar gidebilir. Ense sertliği ve meningeal iritasyon bulguları nadirdir. Davranış-kişilik değişiklikleri gözlenebilir. Konvülziyonlar olabilir, etkilenen bölgeye göre fokal bulgular (paraparezi, hemiparezi, beyinsapı bulguları) görülebilir. Optik nöropati de sıktır. Nadiren ADEM tablosuna periferik sinir sistemi tutulumu (akut inflamatuar demiyelinizan polinöropati) de eşlik edebilir. Klinik olarak ADEM ile MS ayrımında en önemli bulgunun ensefalopati varlığı olduğu ileri sürülmektedir.
Seyir çoğu zaman monofaziktir. Ancak bazen akut atak sırasında bunun monofazik mi olacağı, yoksa yineleyerek multipl skleroz şekline mi dönüşeceğini kestirmek güç olabilir. Bulguların yaygın olması, hastada ensefalopatik bir tablo bulunması daha çok ADEM lehine ise de, multipl skleroz da nadiren bu şekilde karşımıza çıkabilir. ADEM ve MS arasındaki ayrımın dışında son yıllarda bir de yineleyici ADEM (relapsing ADEM) olgularından söz edilmektedir. Büyük serilerde olguların yaklaşık %15-30’unun yineleyici ADEM şeklinde karşımıza çıkabileceği belirtilmektedir. Nükslerin genellikle ilk yıllar içinde görülebildiği bildirilmekle beraber, ilk ataktan yıllar sonra görülebilen olgular da bildirilmiştir. Nüks riskini öngören faktörler arasında optik nörit varlığı, ailede başka demiyelinizan hastalık öyküsü, ilk ataktan sekelsiz düzelme ve MR’da MS kriterlerinin doldurulması sayılmıştır. Bu olguların gerçekten tekrarlayan ADEM hastalığını mı temsil ettikleri, yoksa bir tür yineleyici MS formu mu oldukları halen tartışmalı bir konudur. Çok nadiren erken dönemde yineleyici ADEM gibi seyreden olguların ileriki yıllarda sekonder progresif MS gibi bir klinik seyir gösterebildiği de bildirilmiştir.
Klinik prezentasyon dışında MR bulguları da ayrımda bize yardımcı olabilir. ADEM’de görülen ak madde lezyonları daha yaygın, geniş ve simetrik olma eğilimindedir (Şekil 10). Görülen bütün lezyonların benzer kontrast tutma paterni göstermesi lezyonların aynı yaşta olduğunu gösterir. Yine de bazı lezyonlar kontrast tutarken bazılarının kontrast tutmaması da söz konusu olabilir. MR’da bilateral simetrik paternin görünmemesi, T1 ağırlıklı kesitlerde kara deliklerin görülmesi ve 2 veya daha fazla periventriküler lezyon bulunmasının MS lehine kabul edilebileceği belirtilmektedir. Daha kesin bir ayrım için izleme döneminde ataktan en az üç ay sonra MR kontrolü yapılmalı ve ortaya yeni lezyonların çıkıp çıkmadığı araştırılmalıdır. Yeni lezyon gelişmesi veya üç ay sonraki MR’da kontrast tutan lezyon saptanması tanıyı multipl skleroz lehine çevirecektir. Klinik olarak kesin bir ayrım yapılabilmesi için hastanın en az 6 ay takibinin gerektiğini ileri süren görüşler vardır. Aslında klinik olarak bu ayrımın yapılmasının güç olması kadar, histopatolojik ayrımı da güç olabilir.
Şekil 10. ADEM. Kranyal MR, T2 ağırlıklı aksiyel kesitlerde iki yanlı oldukça simetrik geniş ak madde lezyonları görülmektedir
(Resimler, İstanbul Tıp Fakültesi, Noroloji Anabilim Dalı Multipl Skleroz Birimi materyelinden saglanmıştır).
Patoloji
Başlıca ak maddeyi tutan, ancak gri maddeyi de etkileyen yaygın perivasküler inflamasyon söz konusudur. Hem lenfositler, hem plazma hücreleri hem de makrofajlar bu inflamasyonu oluşturur. Çeşitli düzeylerde demiyelinizasyon da vardır, ancak multipl skleroz ile karşılaştırıldığında bu oldukça geri plandadır. ADEM yelpazesinin diğer ucunda akut hemorajik ensefalomiyelit (Hurst hastalığı) tablosu yer alır. Non-spesifik bir üst solunum yolu infeksiyonundan yaklaşık 10 gün kadar sonra ortaya çıkan çok akut bir ensefalomiyelit formudur. Hızla konfüzyon-koma tablosu gelişir ve sıklıkla fatal seyreder. Nötrofilik pleositoz nedeniyle bakteriyel infeksiyonla karıştırılabilir. Demiyelinizasyon olmaksızın hem gri hem ak maddede perivenüler hücre infiltrasyonu, kanama, ödem ve nekroz görülebilir. Post vaksinial ensefalomiyelit de akut hemorajik ensefalomiyelit ile ADEM arasında değişen spektrumda karşımıza çıkabilir.
Laboratuvar İncelemeleri
ADEM hastalarında kanda saptanabilen spesifik bir bulgu yoktur. Ancak olası öncü infeksiyonlar ve karışabilecek diğer hastalıklar açısından bazı incelemeler yapılmalıdır. Bunlar arasında tam kan sayımı, saatlik sedimentasyon hızı, SLE ve diğer sistemik hastalıkların belirteçleri ve viral serolojik incelemeler sayılabilir. Mikoplazma antikorları ve titrede olası yükselmeler de araştırılmalıdır. BOS’ta lenfositik pleositoz bulunabilir, nadiren hücresiz de olabilir. Protein artmıştır, şeker normal veya hafifçe azalmış olabilir. Oligoklonal bantlar sık olmamakla birlikte görülebilir. Ancak multipl sklerozda görülenin aksine, zaman içinde kaybolması beklenir. Eğer BOS hücre düzeyi doğrudan infeksiyöz bir süreci düşündürürse mutlaka gerekli kültürler yapılmalıdır.
Tedavi
Bugün ADEM tedavisinde ilk seçilen yaklaşım yüksek doz intravenöz metilprednizolon verilmesidir. Genellikle 3-10 gün üstüste 1000 mg/g intravenöz metilprednizolon verildikten sonra hastanın verdiği cevaba göre intravenöz olarak veya oral metilprednisolonla doz yavaş yavaş azaltılır. Diğer bir steroid seçeneği 16-32 mg/g deksametazon verilmesidir. Yakın zamanda yürütülen randomize ve kontrollü bir çalışmada steroide cevapsız olguların plazmafereze iyi cevap verdiği bildirilmiştir. Eğer doğrudan infeksiyöz bir süreç dışlanamamışsa mutlaka uygun antibakteriyel ve antiviral tedaviye de başlanmalıdır.
Miyelini Daha Geri Planda Etkileyen, Ancak Multipl Skleroza Benzer Tablolara Yol Açabilecek Diğer İnflamatuar MSS Hastalıkları
Konnektif Doku Hastalıkları ve Sistemik Vaskülitlere Bağlı Tutulum
Sistemik Lupus Eritematosus (SLE)
SLE tanı kriterleri arasında malar raş, fotosensitivite, oral ülserler, artrit, böbrek tutulumu, hematolojik tutulum, nörolojik tutulum, antinükleer antikor ve diğer immunolojik bulguların varlığı sayılabilir. SLE’de MSS tutulumu görece sık görülse de, genellikle sistemik hastalığın varlığı ayırıcı tanıda kuşkuya yer bırakmaz. Sıklıkla sistemik hastalık önce ortaya çıkar. Nadiren hastalık nörolojik tablo ile başlar. Nörolojik tutulum çeşitli şekillerde karşımıza çıkabilir: hem yaygın MSS bulgularına yol açabilir, hem sınırları belirli fokal tutulum gösterebilir. Yaygın MSS tutulumu psikotik bulguların eşlik edebildiği demansiyel bir tablo şeklinde olabilir. Kognitif bozukluk nörolojik lupusun önemli bir göstergesidir. Sadece nöropsiklojik değerlendirme ile ortaya konabilen ‘sessiz’ tutulumdan ağır demansiyel bir tabloya kadar değişebilir. Hastalığın aktivasyon dönemlerinde uyanıklık kusuru veya deliryum tablosu gelişebilir. SLE hastalarında gelişen major depresyon ve diğer afektif bozukluklar ile akut psikotik tabloların da nörolojik tutulum lehine kabul edilmesi gerektiğini düşünenler vardır. Ensefalopati tabloları dışında SLE seyri sırasında aseptik menenjit veya psödotümör serebri de gelişebilir. Özellikle uzun süren ve progresif seyirli başağrılarında bunlar akla gelmelidir. Antifosfolipid sendromu bulunan SLE hastalarında epileptik nöbetler de görülebilir. Bunlardan başka SLE hastalarında iskemik veya kanayıcı serebrovasküler olaylar, nöbetler, hareket bozuklukları, periferik nöropatiler ve multipl skleroz benzeri klinik tablolar da görülebilir ve internükleer oftalmopleji, paroksizmal tonik spazmlar, optik nöropati, omurilik hastalığı şeklinde nörolojik bulgular olabilir. Lupus hastalarında gelişen görme kaybı retrobulber optik nörit veya iskemik optik nöropatiye bağlı olabilir. SLE’de görülen transvers miyelit tablosunun anti-fosfolipid antikorları ile ilişkili olabildiği düşünülmektedir. Tipik multipl skleroz veya daha sıklıkla nöromiyelitis optika ile SLE birlikteliği gösteren olgular bildirilmiştir.
SLE olgularında nörolojik tutulumun patogenezinde en önemli rolün vasküler faktörlere ait olduğu, bunun altında da ya antifosfolipid antikorların ya da lupus endokarditinin yattığı düşünülmektedir. Bir diğer faktör de otoantikorlara bağlı immunkompleks birikimlerinin yol açtığı vaskülit tablosu veya direkt otoantikorlara bağlı nöronal hasardır. Bütün bunların dışında, lupus ile doğrudan ilişkili olmayan nörolojik olayların da görülebileceği unutulmamalıdır. Bu durumların başında immunsupresif tedaviye bağlı gelişebilecek MSS infeksiyonları gelmektedir. Başka nörolojik bulguların eşlik etmediği epizodik baş ağrılarının lupusun nörolojik tutulumu lehine alınmaması gerektiği unutulmamalıdır.
BOS’ta hücre ve/veya protein artışı saptanabilir; oligoklonal IgG saptanma oranı yaklaşık %40’tır. MR’da hem periventriküler hem uzak ak madde lezyonları görülebilir; multipl sklerozdan ayırdetmek güç olabilir. Eğer arter sulama alanına uyan lezyonlar da eşlik ediyorsa, ayırıcı tanıda yardımcı olabilir. ANA ve diğer otoantikorların saptanması yol gösterici olsa da multipl sklerozda da 1/80- 1/160 gibi görece düşük titrede ANA pozitifliği % 30’u bulabilir. Bakılabilirse, anti-ribozomal P protein antikorlarının psikotik tablo gösteren lupuslularda yüksek olduğu dikkati çekmiştir.
MSS lupusu tedavisi SLE’nin diğer organ tutulumlarındaki gibidir; yüksek doz intravenöz kortikosteroidlerin yanı sıra siklofosfamid gibi diğer immunsupresif ajanlar da verilir.
Sjögren Sendromu
Sjögren sendromu, eksokrin bezlerin lenfositik infiltrasyonu ile giden otoimmun bir hastalıktır. Keratokonjonktivitis sicca, kserostomi ve bir diğer bağ dokusu hastalığı (sıklıkla romatoid artrit) triadı ile karakterizedir. Daha çok orta yaştaki kadınlarda görülür. Hastaların dörtte birinde deri döküntüsü, interstisyel nefrit, lenfadenomegali gibi sistemik bulgular da vardır; lenfoma ile ilişkisi bilinmektedir.
Genellikle periferik sinir sistemini tutarsa da MSS tutulumu olan olgular da bildirilmiştir. Görülen nörolojik tablolar arasında epileptik nöbetler, inme benzeri yerleşen fokal nörolojik bulgular, yaygın meningoensefalit tabloları yer alır. Bunlar dışında miyelopati, optik nöropati, Devic sendromu ile birlikte görülen veya yineleyici multipl sklerozu taklid eden olgular da bildirilmiştir. Özellikle son yıllarda Sjögren sendromu ile nöromiyelitis optika arasındaki ilişki giderek daha fazla dikkati çekmektedir. Bu olgularda eşlik edebilen periferik nöropati veya miyozit gibi multipl skleroz hastaları için atipik sayılabilecek bulgular da bulunabilir.
Genellikle Sjögren sendromu tanısı nörolojik tablonun ortaya çıkışından daha sonra koyulursa da iyi bir sorgulama sistemik belirtilerin önceden beri bulunduğunu ortaya koyabilir. Anti-Ro ve anti-La gibi otoantikorların yanı sıra diğer pek çok otoantikor da bulunabilir (anti-dsDNA, anti-histon, anti-Sm, romatoid faktör gibi). BOS’ta monoklonal veya oligoklonal IgG sıklıkla pozitiftir. MR bazı olgularda multipl sklerozdakine benzer lezyonlar ortaya koyar. Ancak bu lezyonların demiyelinizan natürlü olmadığı, vaskülitik lezyonlar olabileceği düşünülmektedir. Tanı için gözyaşı sekresyonunu ölçen Schirmer testi yeterli olabileceği gibi, tükrük bezi biyopsisi gerekebilir.
Sistemik Skleroz
Cilt ve diğer dokularda aşırı kollajen birikimi ile karakterize, otoimmun natürlü olduğu düşünülen bir hastalıktır. Skleroderma bulgusuna sıklıkla Raynaud fenomeni, kalsinoz, sklerodaktili, disfaji, telanjiektazi eşlik eder. İç organlar tutulabilir. Genellikle periferik sinir sistemini tutar. Kranyal sinirlerin tutulumu da bildirilmiştir. Bir de vaskülite bağlı MSS bulguları ortaya çıkabilir. Bazı olgularda miyelopati ile giden multipl skleroz benzeri tablolar bildirilmişse de arada bir ilişki olup olmadığı net değildir.
PAN, Wegener ve Diğer Sistemik Vaskülitler
Vaskülitler damar duvarının inflamatuar infiltrasyonu ve nekrozu ile giden ve birbirinden çok farklı tabloları kapsayan bir hastalıklar grubudur. Genellikle tuttukları damar çapına göre sınıflanırlar. Büyük arterleri tutan belli başlı iki vaskülit vardır: dev hücreli arterit (temporal arterit) ve Takayasu arteriti. Bunlarda sinir sistemi tutulumu daha çok inme benzeri tablolarla karşımıza gelir (Bakınız:Gençlerde inme). Klasik poliarteritis nodosa (PAN) ve Kawasaki hastalığı, orta boy arterleri tutar. Wegener hastalığı, Churg-Strauss ve mikroskopik polianjiit ise orta boy ve daha küçük arterleri tutar. Sadece küçük damarları tutan vaskülitler ise Henoch-Schonlein purpurası, esansiyel kriyoglobulinemi ve cildin lökositoklastik vaskülitidir. Bir de çeşitli infeksiyonların seyri sırasında gelişen nadir vaskülit tabloları vardır.
Sistemik vaskülitlerin hemen hemen bütün tipleri merkezi veya periferik sinir sistemi tutulumuna yol açabilir. Ancak bir de merkezi sinir sisteminin izole vasküliti ve periferik sinir sisteminin izole vasküliti (non-sistemik vaskülitik nöropati) olarak adlandırılan, sistemik tutulumun olmadığı tablolar vardır. Bunlardan ilki bir sonraki başlık altında, ikincisi ise periferik sinir hastalıkları bölümünde anlatılmaktadır.
Sistemik vaskülitlerin seyri sırasında ortaya çıkan MSS tutulumunda genellikle diğer organ sistemlerine ait bulgular mevcut olduğundan tanınmaları zor olmaz. Ancak nadiren sistemik tutulum geri planda olabilir veya hastalık MSS tutulumu ile ortaya çıkmış olabilir. Bu durumda tanı koyma görevi nörologa düşebilir. Klinik tablo tek veya multipl iskemik inme ile karsımıza çıkabildiği gibi, yaygın bir ensefalopati tablosu da görülebilir. Bu durumda uyanıklık kusuru, nöbetler, progresif davranış ve kişilik değişikliği, hareket bozuklukları, beyinsapı bulguları görülebilir. Eşlik eden periferik sinir tutulumu da varsa PAN, Wegener hastalığı, Churg-Strauss ve esansiyel kriyoglobulinemi akla gelmelidir.
Semptomatik olmasa da diğer organ tutulumları araştırılmalıdır. Böbrekler, akciğerler, paranazal sinüsler, ülseratif ve diğer tipte deri lezyonları araştırılmalıdır. Örneğin Wegener hastalığında sık görülen triad sistemik arterite eşlik eden üst ve alt solunum yollarında nekrotizan granülomlar ve granülonefrit tablosudur; buna karşılık Churg-Strauss hastalığında ateş, astım, dokularda ve kanda eozinofili ile giden sistemik arterit söz konusudur. Klasik PAN olgularında ateş, halsizlik ve hipertansiyon gibi sistemik yakınma ve bulguların yanı sıra böbrek, deri ve gastrointestinal sistem tutulumu da görülebilir; bu olgularda periferik sinir tutulumu %75’leri bulurken MSS tutulumu çok daha seyrektir (%3-20 arası). Mikroskopik polianjiit PAN’a benzer, ancak ek olarak hızlı ilerleyici glomerülonefrit ve akciğer tutulumu vardır; periferik nöropati çok daha seyrek görülür.
Laboratuvar incelemelerinden saatlik sedimentasyon hızı, tam kan sayımı, kan biyokimyası ve idrar analizi bilgi verici olabilir. CRP, kompleman düzeyleri, kriyoglobulinler ve serolojik testlerden ANA, romatoid faktör (RF), antinötrofil sitoplazmik antikorların (ANCA) alt grupları, anti-Ro ve anti-La, antikardiyolipin antikorlar araştırılmalıdır. ANCA antikorlarının sitoplazmik alt grubu (cANCA) Wegener hastalığı ile ilişkilidir; perinükleer alt grubunun (pANCA) ise daha çok mikroskopik polianjiit ve Churg-Strauss hastalığı ile ilişkili olduğu düşünülmektedir. Gereken durumlarda cilt, kas, periferik sinir veya nazal sinüs biyopsisi yapılabilir. EMG sessiz kalmış periferik sinir tutulumunu ortaya koyabilir. Kranyal MR ve anjiografi MSS tutulumu ile ilgili bilgi verir.
Sistemik vaskülitlerin prognozu çok yüz güldürücü değildir. Bu nedenle tanı koyulur koyulmaz yüksek doz steroid ve diğer immunsupresif ajanlarla tedaviye başlanmalıdır.
Primer MSS Vasküliti
Primer MSS vasküliti veya diğer adıyla MSS’nin granülomatöz anjiiti, çok ender rastlanan bir hastalıktır. Sistemik vaskülitlerde olduğu gibi MSS’ye ait fokal veya jeneralize belirti ve bulgularla ortaya çıkabilir. Genellikle akut veya subakut bir seyir gösterse de bazen kronik progresif gidişli olabilir. Başağrısı oldukça sık rastlanan bir bulgudur. Progresif olarak artabilir, kusma ve ense sertliği eşlik edebilir. Davranış ve kişilik değişikliği, kognitif bozukluklar da sıktır. Uyanıklık kusuru gelişebilir, nöbetler olabilir. Sistemik vaskülitlerde sıkça görülen inme benzeri fokal tablolar burada daha geri plandadır. Nadiren de klinik tablo kitle lezyonu veya kronik menenjit şeklinde karşımıza çıkabilir.
Sistemik vaskülitlerin aksine kan sayımı ve sedimentasyon hızı ılımlı değişiklikler dışında normaldir. BOS genellikle patolojiktir. Birkaç yüze varan lenfositik pleositoz ve protein artışı gözlenebilir. Kanda yukarıda sayılan serolojik testlerin yanı sıra infeksiyon ajanlarına yönelik serolojik incelemeler, BOS kültürü ve sitolojisinin yapılması diğer olasılıkların dışlanması için gereklidir.
EEG’de sıklıkla yaygın yavaşlama görülür, ancak bu spesifik bir bulgu değildir. Kranyal MR ile hemen daima bazı patolojik bulgular görülür, ancak spesifik değildir. Serebral anjiografi ile orta ve küçük boy arterlerde daralmalar ve düzensizlikler görülebilirse de hem yalancı pozitiflik oranı hem de yalancı negatiflik oranı yüksek bir inceleme yöntemidir. Tanıda beyin ve meninks biyopsisi ‘altın standart’ olarak kabul edilirse de invazif bir yöntem olan biyopsinin de yalancı negatiflik riski vardır. Eğer MR’da görülen bir lezyon varsa buradan biyopsi alınması pozitiflik şansını arttıracaktır.
Primer MSS vasküliti çok ağır seyreden ve fatal olabilen bir hastalıktır. Bu nedenle tanı koyulur koyulmaz tedaviye başlanmalıdır. Seyrek görülen bir hastalık olduğu için prospektif tedavi çalışmaları yoktur. Ancak, yüksek doz steroid ve diğer immunsupresif ajanlar kullanılmaktadır. Bugün pek çok merkezde geçerli olan uygulama yüksek doz intravenöz metilprednizolon verilmesi, buna rağmen progresyon sürerse oral veya intravenöz siklofosfamid eklenerek en az bir yıl sürdürülmesidir.
Behçet Hastalığı* ve Sinir Sistemi Tutulumu
Behçet hastalığı, yineleyici oral aftlar, genital ülserasyonlar ve uveit ile kendini gösteren multi-sistemik inflamatuar bir hastalıktır. Bazı olgularda oral, genital mukoza ve uvea dışında eklemler, damarlar (özellikle venler), gastrointestinal sistem, akciğerler ve sinir sistemi gibi sistemlerin tutulumu da görülebilir. Behçet hastalığında sinir sistemi tutulumu olguların yaklaşık % 5-10’unda görülür. Bu olguların hemen hepsinde merkezi sinir sistemi etkilenir; buna karşın, periferik sinir ve kas tutulumu oldukça seyrektir. 1990 yılında yayınlanan uluslararası tanı kriterlerine göre, Behçet hastalığı tanısı için oral aftların yanı sıra aşağıdakilerden en az ikisi bulunmalıdır: genital ülserasyon veya skarı, deri lezyonları (akne, folikülit, eritema nodozum veya genital bölge dışında ülserasyonlar), göz tutulumu (bir göz hekimi tarafından saptanan ön veya arka uveit ya da retinal vaskülit) ve pozitif paterji testi.
*Profesör Dr. Hulusi Behcet : 20 Şubat 1889’da, Istanbul’da doğdu. 1910 yılında Gülhane Askeri Tıp Fakültesi’nden mezun oldu. Ardından Dermatoloji ve Sifiloloji Bölümü’nde ihtisas yaptı. I.Dünya Savaşı sırasında çeşitli askeri hastanelerde görev yaptı. Daha sonra bir yıl süre ile Berlin ve Budapeşte’de dermatoloji alanında çalıştı. Türkiye’ye döndüğünde önce Hasköy Zührevi Hastalıklar Hastanesi’nde başhekim, daha sonra da Guraba Hastanesi’nde uzman olarak çalıştı; 1933 Üniversite Reformu ile İstanbul Üniversitesi-İstanbul Tıp Fakültesi’ne bağlanan bu kurumda profesörlüğe ve kürsü direktörlüğüne atandı.
1924 yılında oral aftları, genital ülserleri, eritema nodosum, ve görme kaybı bulunan ilk olgusunu görmüştür. Önceden tüberküloz veya sifiliz tanıları almış olan bu olguyu yıllarca izler, ancak onun tanısı bir viral infeksiyondur. Benzer bir klinik tablo gösteren diğer iki olguyu da 1930 ve 1936 yıllarında görmüştür. Bu gözlemlerini ilk kez 1937 yılında yayınlamıştır.
1936 yılında Dermatologische Wochenschrift ve Medizinische Welt gibi dergilerin yayın kuruluna davet edilmiştir. Fransız, Alman, Avusturya, Macaristan ve Yunanistan Dermatoloji Topluluklarının üyesi olmuştur. 8 Mart 1948 tarihinde kalp krizi nedeniyle 59 yaşında yaşama veda etmiştir.
Klinik Tablo
Genelde Behçet hastalığı erkeklerde daha sık gorülür; nörolojik tutulum söz konusu olduğunda erkekler lehine olan bu dağılım daha da belirginleşir. Çoğu zaman nörolojik tutulum Behçet hastalığının başlangıcından birkaç yıl (ortalama 5 yıl) sonra ortaya çıkar ve başlıca iki ana şekilde görülür. MSS parenkiminin tutulduğu durumlarda klasik tablo birkaç hafta içinde progresif olarak yerleşen davranış değişikliği, hemiparezi, ataksi ve dizartri ile karakterize bir beyinsapı sendromudur. Bazen yüksek ateş ve başağrısı, nadiren de menenjizm eşlik edebilir. Olguların önemli bir kısmında sfinkter kusuru bulunabilir. Duyusal yakınma ve bulgular seyrek görülür. Bazı olgularda ise omurilik tutulumu ön planda olabilir. Bunun dışında bir de MSS parenkiminin tutulmadığı, ancak majör damarlarının tutulumuna bağlı olarak nörolojik bulguların ortaya çıktığı ikinci bir grup hasta vardır. Bu grupta en sık serebral venöz sinüslerin tıkanması görülür ve klasik kafa içi basınç artışı sendromu ile karşımıza gelir. Çok daha seyrek olarak da serebral arterlerin tıkanmasına bağlı inme-benzeri tablolar görülebilir.
BOS Bulguları
Olguların yaklaşık dörtte üçünde BOS bulguları saptanabilir; milimetreküpte birkaç yüze kadar, bazen 1000 kadar hücre bulunabilir. Hem nötrofilik hem de lenfositik hakimiyet söz konusu olabilir. BOS proteininde ılımlı bir artış söz konusudur (genellikle <100 mg/dl); şeker düzeyi normaldir. BOS’ta ,IgG düzeyi artmıştır; ancak oligoklonal IgG bantları seyrek bulunur. Dural sinüs trombozu bulunan olgularda açılış basıncı yüksekliği dışında BOS genellikle normaldir.
Nöroradyolojik Bulgular
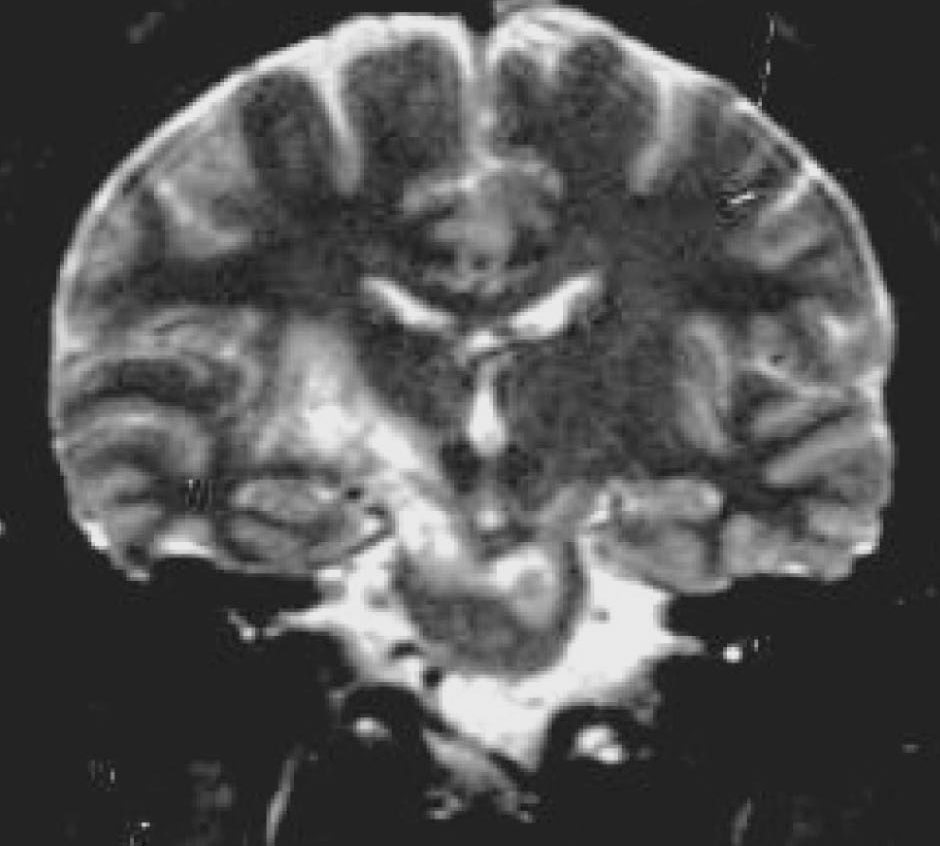
Behçet hastalığında seçilecek yöntem MR’dır. Akut atak sırasında kranyal MR bulguları oldukça tipiktir: genellikle beyinsapından aynı tarafta diensefalik yapılara ve bazal ganglion bölgesine uzanan geniş bir lezyon görülür. Bazen iki yanlı da olabilir (Şekil 11). Daha sonra bu lezyon küçülür, dağınık noktasal lezyonlar haline dönüşür veya tamamen kaybolabilir. Geç dönemde beyinsapı ve diensefalik yapılarda bazen asimetrik olabilen bir atrofi görülebilir.
Şekil 11. NöroBehçet. Kranyal MR, koronal T2 ağırlıklı kesitlerde sağda bazal ganglionlar bölgesinden talamusa ve iki yanlı mezensefalona ulaşan, solda ponsa da uzanan hiperintens lezyon görülmektedir (Kaynak 1’den alınmıştır).
Tedavi
Akut atak tedavisinde yüksek doz intravenöz (1000 mg/gün, genellikle 5 gün boyunca) metilprednizolon pulse tedavi verilir. Daha sonra oral idame tedavisine geçip dozu çok yavaşça azaltmak gerekir. Ani kesilme durumunda tedaviye daha dirençli nüksler görülür. Uzun vadeli koruyucu tedavi olarak da intravenöz steroidle beraber 2.5 mg/kg/gün dozunda azathioprin’e başlamak yararlı olabilir. Azathioprin tedavisinin süresi ile ilgili bir fikir birliği yoksa da en az birkaç yıl sürdürülmesi gerektiği kabul görmektedir. Bazen akut atak sırasında steroide cevap yetersizse aylık pulse intravenöz siklofosfamid eklenebilir. Tek tek olgu örneklerinde TNF antagonistlerinin de yararlı olduğunu bildiren çalışmalar yayınlanmıştır. Siklosporin almakta iken nörolojik tutulum gelişen olgularda siklosporinin kesilmesi ve o hastada tekrar başlanmaması gerekmektedir. Dural sinüs trombozu tedavisinde de başlıca kullanılan ajanlar steroidlerdir. Beraberinde antikoagulan tedavi verilmesinin gerekip gerekmediği tartışmalı bir konudur. Antikoagülan tedavi verilecekse mutlaka öncesinde toraks BT yapılarak pulmoner anevrizma varlığı dışlanmalıdır.
Seyir ve Prognoz
Behçet hastalığının diğer bulguları gibi, nörolojik tutulum da ataklar ve sekelli/ sekelsiz düzelmelerle gider. Ancak küçük bir grup hastada herhangi bir atak olmaksızın başlangıçtan itibaren ilerleyici bir seyir söz konusudur. Ataklı hastaların ise yaklaşık yarısında daha sonra progresif bir kötüleşme görülebilir. Behçet hastalığında nörolojik tutulum olması prognozu kötüleştiren bir faktördür. Ancak nörolojik tutulumun tipine göre bazı farklılıklar sözkonusudur. Parenkimal MSS tutulumu içinde kognitif tutulumla birlikte giden yaygın beyinsapı sendromu kötü prognoz işaretidir. Ayrıca, akut atak sırasında BOS patolojik ise bu da uzun vadede kötü prognozu gösterir. Buna karşın, Behçet hastalığı ile ilişkili dural sinüs trombozunun prognozu gerek parenkimal tutuluma göre, gerekse diğer nedenlere bağlı sinüs trombozlarına göre çok iyidir.
Nöro-Sarkoidoz
Sarkoidoz etyolojisi bilinmeyen multisistemik bir granülomatöz hastalıktır. Genellikle genç erişkinlerde görülür ve kadınlarda biraz daha sıktır. Başlıca akciğerleri tutar, sinir sistemi tutulumu olguların yaklaşık %5’inde görülür. Sarkoidoz, tanısı güç bir hastalıktır; çoğu zaman doku biyopsisine dayanır. Tutulan dokularda önce CD4+ lenfositler ve mononükleer fagositler toplanır, giderek makrofajların, epiteloid hücrelerin ve bunların oluşturduğu dev hücrelerin birikimi ile granülomlar gelişir. Bu granülomun dış çevresinde CD4+ lenfositler dizilir. Granülomun ortasında kazeifikasyon görülmez.
Klinik Özellikler
Sıklıkla aylar içinde yavaş progresif bir tablo olarak karşımıza çıkar; akut veya subakut formları olabileceği gibi, asemptomatik hastalar da bildirilmiştir. Genellikle ateş, halsizlik, iştahsızlık, kilo kaybı gibi belirtilerle ortaya çıkar. Daha sonra tutulan organa göre bulgular verir. Olguların %90’ında hastalığın bir döneminde akciğer tutulumu görülür. Bu durumda dispne, kuru öksürükle kendini gösterir; hemoptizi, balgam nadirdir. Plevra tutulumu da seyrek görülür (< %5). Gerek toraks içinde gerekse diğer bölgelerde lenfadenomegali de sık görülür. Cilt tutulumu yaklaşık %25 olguda söz konusudur; eritema nodozum, makülopapüler döküntü ve ciltaltı nodülleri gelişebilir. Yine %25 olguda uveit görülebilir. Ayrıca kemik kistleri, eklem tutulumu ve asemptomatik miyozit görülebilir. Karaciğer tutulumu sıktır, ancak nadiren klinik bulgulara yol açar; böbrek, gastrointestinal kanal ve kalp tutulumu nadir görülür.
Sarkoidoz seyrinde görülebilecek iki sendrom tanımlanmıştır: Löfgren sendromu, eritema nodozum, bilateral hiler lenfadenomegali, eklem tutulumu ile karakterizedir; Heerfordt-Waldenström sendromunda ise ateş, uveit, parotis tutulumu ve yüz felci görülür.
Nörolojik tutulum-klinik özellikler: Bir seride görülen klinik tabloların dağılımına göre, en sık rastlanan bulgu subakut seyirli atipik özellikler gösteren optik nöropatidir. Olguların üçte birinde iki yanlı olabilir. İkinci sıklıkta kranyal sinir tutulumu gelir; bu grupta en sık VII. sinir, ardından VIII. sinir, oküler motor sinirler ve V. sinir tutulumu görülür. Yüz felci yineleyici olabilir. Üçüncü sırada omurilik tutulumu, ardından da beyinsapı ve serebellum tutulumu gelir. Az sayıda olguda kognitif bozukluk, menenjizm, hidrosefali, veya hipotalamik disfonksiyon görülür.
Tanıda Yardımcı İnceleme Yöntemleri
Yukarıda da belirtildiği gibi, sarkoidoz tanısı histopatolojik doğrulamaya dayanır. Bu nedenle hastada MSS dışında organ tutulumu varsa buradan biyopsi materyeli alınabilir. Sistemik sarkoidoz açısından toraks grafisi veya BT yapılmalıdır. Bilateral hiler lenfadenomegali, yaygın interstisyel tutulum paterni veya ikisinin kombinasyonu görülebilir. Bu şekilde transbronşiyal biyopsi yapılacak bölge hakkında da bilgi elde edilebilir. Bronkoalveoler lavaj da hem diğer olasılıkların araştırılması hem de sarkoidoz tanısının doğrulanması için yol göstericidir. Genellikle artmış CD4+ lenfosit sayısı ve bol miktarda alveolar makrofaj icerir. Galyum 67 ile akciğer ve tüm vücut taraması da sıklıkla pozitif sonuç verir, ancak spesifik değildir. Hastaların yaklaşık üçte ikisinde kanda anjiotensin ‘converting’ enzim (ACE) düzeyi artar, ancak diğer granülomatöz hastalıklarda da pozitif olabilir. İdrarda 24 saatlik kalsiyum atılımının artması da sarkoidoz lehine bir bulgudur, ancak spesifik değildir. Bütün bunların dışında, Kveim-Siltzbach deri testi artık çok uygulanmayan bir yöntem olsa da oldukça duyarlı ve spesifik bir testtir. Ancak hem bekleme süresinin birkaç hafta gibi uzun bir dönem olması, hem güvenliğe ilişkin kuşkular nedeniyle pek çok ülkede kullanılmamaktadır. Sarkoidoz tanısının çoğu zaman bir dışlama tanısı olduğu, hiçbir laboratuvar bulgusunun spesifik olmadığı unutulmamalıdır.
Nöro-sarkoidoz tanısı da benzer derecede güçtür. 1997 yılında yayınlanan bir tanı kriteri önerisine göre, kesin nöro-sarkoidoz tanısı için bu tanıyı düşündüren ve başka bir açıklaması olmayan klinik tabloya eşlik eden pozitif sinir sistemi patolojisi gerekmektedir. Eğer sinir sistemi patolojisi ortaya konmamışsa ancak başka açıklaması olmayan inflamatuar bir sinir sistemi hastalığı varsa ve sistemik sarkoidoza ait patolojik veri varsa olası (probable) nöro-sarkoidoz tanısı söz konusu olmaktadır. Bu kriterin de karşılanmadığı durumlarda ancak muhtemel (possible) nöro-sarkoidoz’dan sözedilebilir.
BOS: Nöro-sarkoidoz hastalarının çoğunda BOS patolojiktir. En sık rastlanan bozukluk protein artışıdır; bazen çok yüksek düzeylere varabilir. Hastaların yaklaşık yarısında lenfositik pleositoz görülebilir. Oligoklonal bantlar saptanabilir. BOS ACE düzeyinin tanıda şimdilik pek yeri yoktur.
Nöroradyolojik incelemeler: Kranyal MR’da kontrast tutulumu da gösteren multipl hiperintens ak madde lezyonları görülür. Meninkslerde kontrast tutulumu da sık görülen bir bulgudur. Bazen bitişik kortekste de hiperintensite veya kontrast tutulumu görülebilir. Omurilik tutulumu olan olgularda ilgili segmenti tutan hiperintens nodüler lezyon görülebilir.
Tedavi:
Genellikle seçilen tedavi yüksek doz oral veya intravenöz pulse metilprednizolon tedavisidir. Bu tedaviye cevap alınamazsa siklofosfamid gibi daha güçlü immunsupresan ajanlar denenebilir.
Kaynaklar
1. Akman-Demir G, Serdaroğlu P, Taşçı B and the neuro-Behçet Study Group. Clinical patterns of neurological involvement in Behçet’s disease. Brain 1999; 112: 1271-1281.
2. Aminoff M (editör). Neurology and General Medicine. 2008 (4. baskı). Churchill Livingstone, Philadelphia
3. Alper G, Heyman R, Wang L. Multiple sclerosis and acute disseminated encephalomyelitis diagnosed in children after long-term follow-up: comparison of presenting features. Dev Med Child Neurol 2009; 51:480-6.
4. Belman AL, Chitnis T, Renoux C, Waubant E; International Pediatric MS Study Group. Challenges in the classification of pediatric multiple sclerosis and future directions. Neurology 2007; 68: S70-4.
5. Bernard G, Riou E, Rosenblatt B, ve ark. Simultaneous Guillain-Barré syndrome and acute disseminated encephalomyelitis in the pediatric population. J Child Neurol 2008; 23: 752-7.
6. Compston A, Ebers G, Lassmann H, Mc Donald I, Matthews B, Wekerle H (editörler). Mc Alpine’s Multiple Sclerosis, 3rd edition. Churchill Livingstone, London, 1998.
7. Cook SD (Editör). Handbook of Multiple Sclerosis. 3rd ed. Marcel Dekker, New York, 2001.
8. Dale RC. Acute disseminated encephalomyelitis: where does it start and where does it stop? Dev Med Child Neurol 2008; 50:326-7.
9. Dean G, Aksoy H, Akalın T, Middleton L, Kyriallis K. Multiple sclerosis in the Turkish and Greek-speaking communities of Cyprus. A United Nations (UNHCR) Bicommunual project. J Neurological Sci 1997;145:163-168.
10. Dilşen N. Behçet hastalığının tarihçesi. Aktüel Tıp Dergisi 1997; 2: 62-65
11. Diker MH. Lavhavi Tasallup.Asabiye Hastalıkları,İkinci cilt, Devlet Matbaası, İstanbul, 1929,;142-156.
12. Hensiek AE, Seaman SR, Barcellos LF, Oturai A, Eraksoy M ve ark. Familial effects on the clinical course of multiple sclerosis. Neurology 2007 , 68:376-383
13. Eraksoy M, Kurtuncu M, Akman Demir G ve ark. A whole genome screen for linkage in Tukish multiplex families with multiple sclerosis. J Neuroimmunol 2003;143; 17-24
14. Eraksoy M. Multipl skleroz’un genetiği. Türkiye Klinikleri 2004, 2:166-17016.
15. Eraksoy M, Yapıcı Z, Akman-Demir G, Ayta S, Özcan H. Tumor-like demiyelinating lesions of the brain and spinal cord in childhood. Mult. Sclerosis 2009;15( suppl 2): S38,P163.
16. Eraksoy M,Yapıcı Z, Akman-Demir G, Kürtüncü M, Özcan H. Children (<10 yrs) with clinically isolated syndrome: long term follow-up for a second clinical event. J Neurol 2008; (suppl 2),O226.
17. Eraksoy M,Yapıcı Z, Akman-Demir G, Ayta S, M, Özcan H. Clinical and laboratory findings in children with neuromyelitis optica : a long-term follow-up J Neurol 2009; (suppl 2): O36
18. Eraksoy M, Akman-Demir G, Kıyat-Atamer et al: The familial occurence of multiple sclerosis in Turkish population. Multiple sclerosis 1998;4:2098.
19. Eraksoy M, Turan N, Kürtüncü M et al:Demographic and clinical findings in familial multiple sclerosis:a hospital based study ( abstact) J Neurol 2002;249(Suppl): 1/113p.430
20. Eraksoy M.Hensiek A,Kurtuncu M, Akman-Demir ve ark. A whole genome screen for linkage in Turkish multiple sclerosis. J Neuroimmunol 2003; 143:129-32.
21. Lublin F, Miller AE. Multiple sclerosis and other inflammatory demyelinating diseases of the central nervous system. Bradley WG, Daroff RB, Fenichel GM, Jankovic J ( editör) Neurology in Clinical Practice. Fifth Edition,Butterworth Heinnemann Elsevier 2008.pp. 1583-1621
22. McDonald WI, Compston A, Edan G, ve ark. Recommended diagnostic criteria for multiple sclerosis: guidelines from the International Panel on the diagnosis of multiple sclerosis. Ann Neurol. 2001; 50: 121-7.
23. Mutlu N. Multiple Sclerosis in Turkey (Etiologic and symptomatologic study of four hundred ten cases) Arch Neurol Psychiatr. 1954,55:511-516.
24. Pastores GM, Kolodyn E.H. Lysosomal Storage diseases . Swaiman E.D., Ashwal S., Ferrıerro D.M. eds Pediatric Neurology , Principles & Practice ,Fourth edition, Mosby Elsevier, Philedelphia,2006 Ch 27, pp 659-714).
Merkezi Sinir Sisteminin Miyelin Hastalıkları >>>





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}